| Version 55 (modified by , 16 years ago) (diff) |
|---|
SNP calling pipeline
Status: Alpha Authors: Freerk van Dijk, Morris Swertz
This is the documentation of the BBMRI-NL snp calling pipeline based on the Broad GATK. It consists of the following three workflows:
- Workflow 1: SnpCallingPipeline/ReferencePreparation
- Workflow 2: SnpCallingPipeline/AlignmentAndCleaning
- Workflow 3: SnpCallingPipeline/VariantCalling
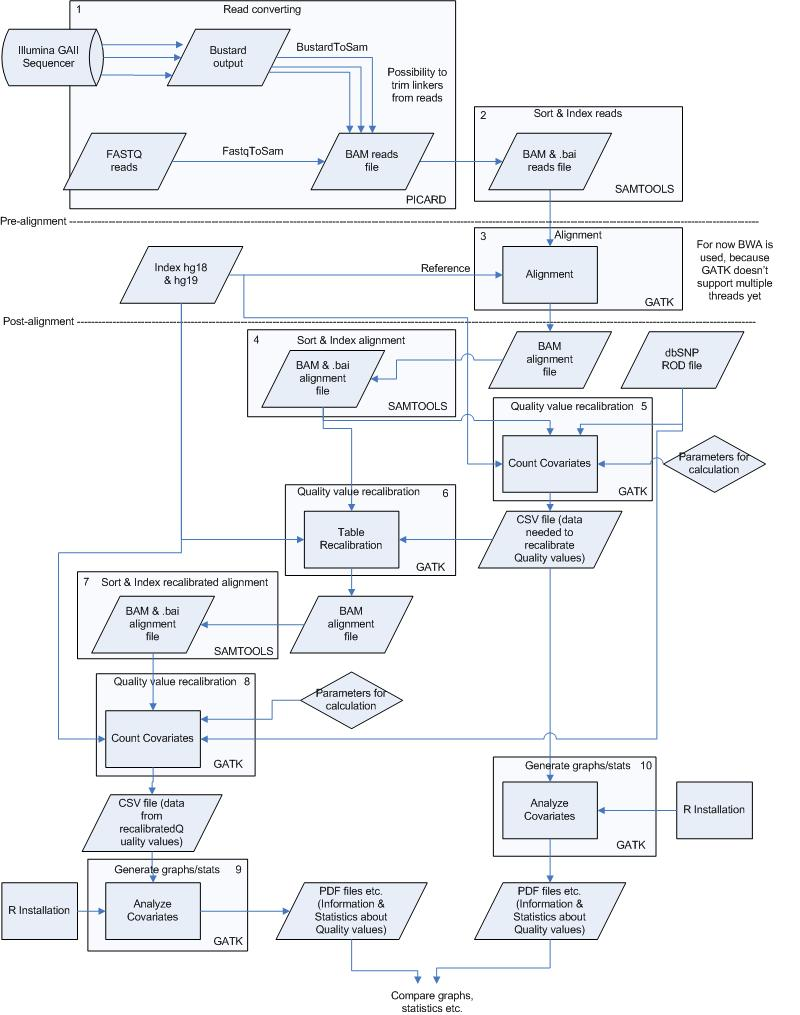
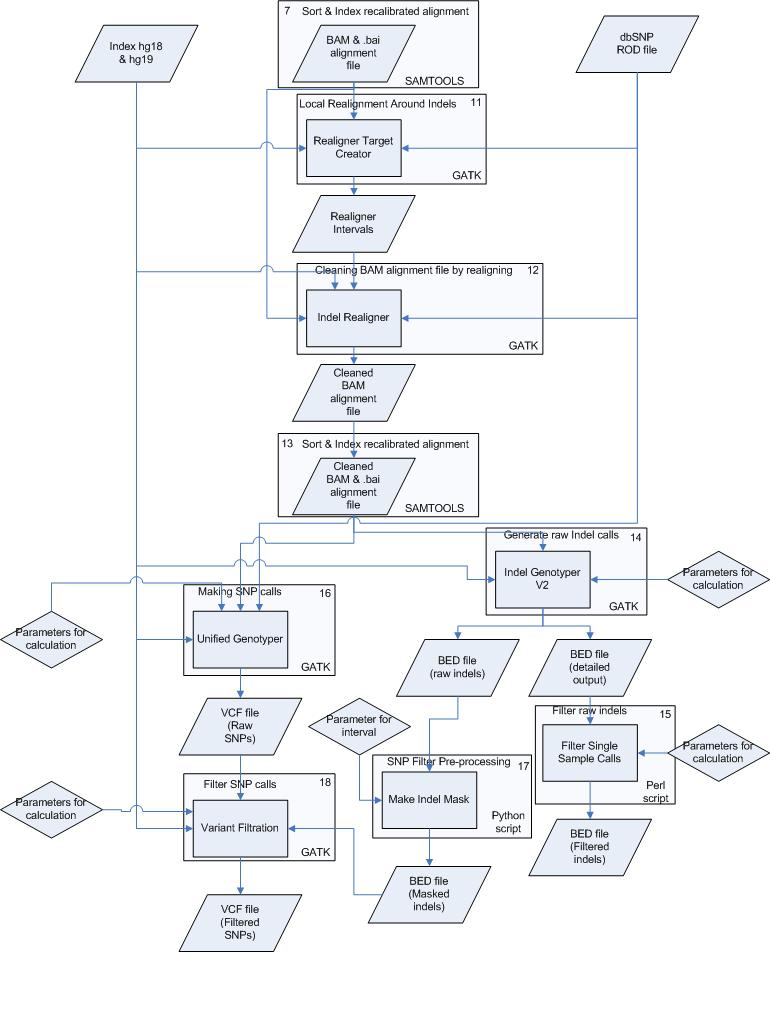
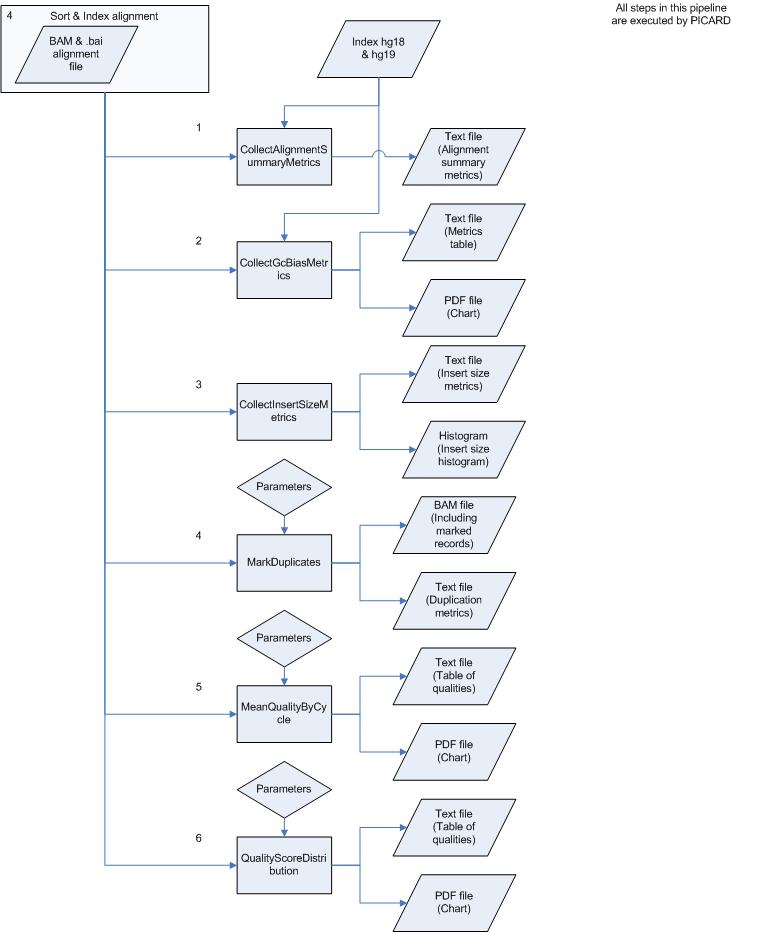
Schematic Overview
This simplified overview this schema hides intermediate sort and indexing steps and only shows data inputs/outputs first time they occur.
Discussion
- How long takes alignment per genome?
- If this takes very long we can split read files
- How long takes realign knownsonly (per genome)?
- If very long, we need to rewrite workflow 2 to split before realign
- For realign: if we split per chromosome, can we also split bam file?
- How to easily lift over from b36 to b37
- Contact BGI if they can use b37??
Todo:
First:
- Recode workflow 2 to work per genome instead of per chromosome and test - Freerk
- Run on pilot data (6) to evaluate timing and concurrency issues (can 6 run on one node?) - Freerk
- Complete analysis of data (60) until including merge to sample.aligned.bam - Freerk
- QC pipeline - Can we get Jeroen and Yurii involved here
- UnifiedGenotyper? without realign - Freerk
- GATK variant eval to make venn diagrams
- Contact Yurii for this; Let Jeroen take charge?
- Share data with Grid following plan Silvia - Freerk
- Contact BGI for sample list - Morris
- Put report on FTP - Ger,Freerk
Next:
- Short tutorial howto generate pipeline scripts - Morris
- Teach Barbara and Jeroen
- Port pipeline to Grid with help of Barbara
- What do we need to generate exactly - Barbara
List of steps
Attachments (3)
- Figure1.png (349.2 KB) - added by 16 years ago.
- Figure2.png (311.5 KB) - added by 16 years ago.
- Figure3.png (224.0 KB) - added by 16 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip