| Version 3 (modified by , 16 years ago) (diff) |
|---|
Table of Contents
Coverage Analysis Pipeline
TODO. Suggested parties to take this up: Antoine van Kampen, Barbera van Schaik, Silvia D Olabarriaga, Mark Santcroos, AMC
Workflows
Create grid directory and change permissions

- Creates a directory on the LFC
- Changes the permissions such that it is in-accessible to the group and others
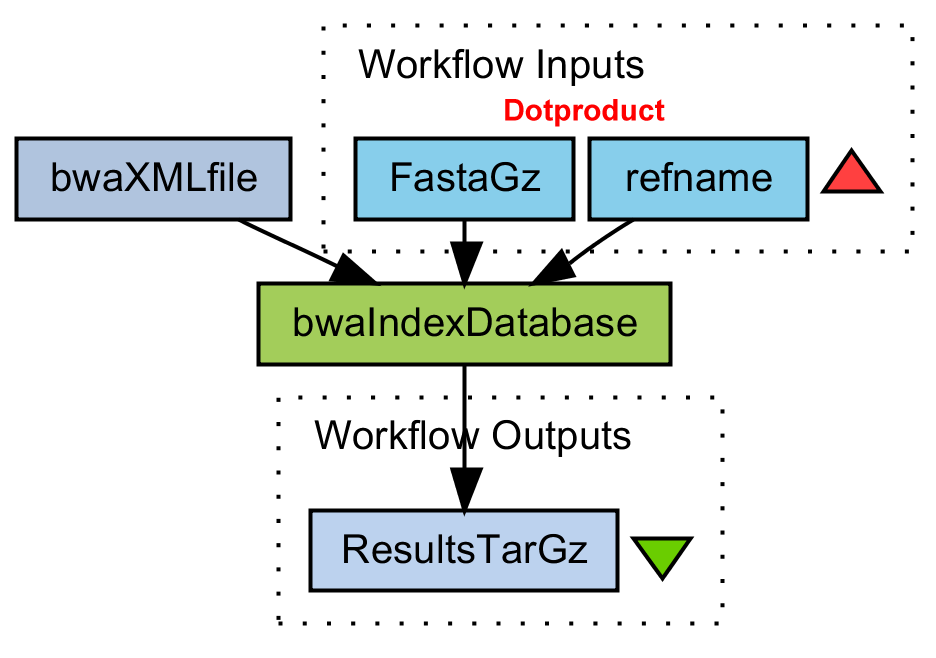
Create a BWA index on database

Gunzip fasta file. Build BWA index. Tar-gzip the results.
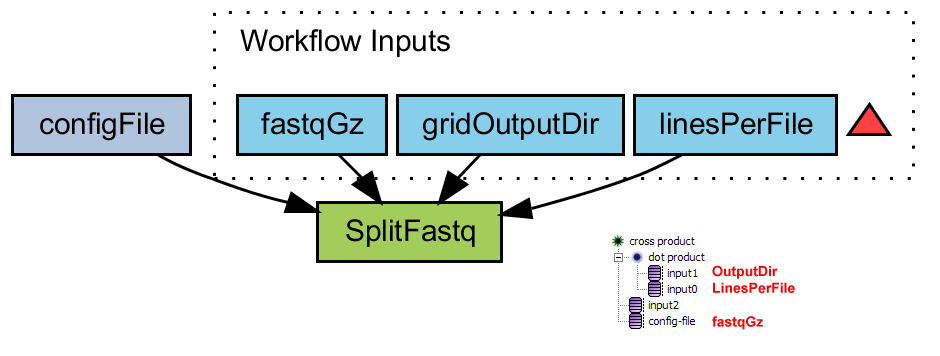
Split fastq file

Splits a large fastq file (gzipped) into several smaller files with the unix command 'split'. The results are uploaded to the directory that is specified in 'gridOutputDir'
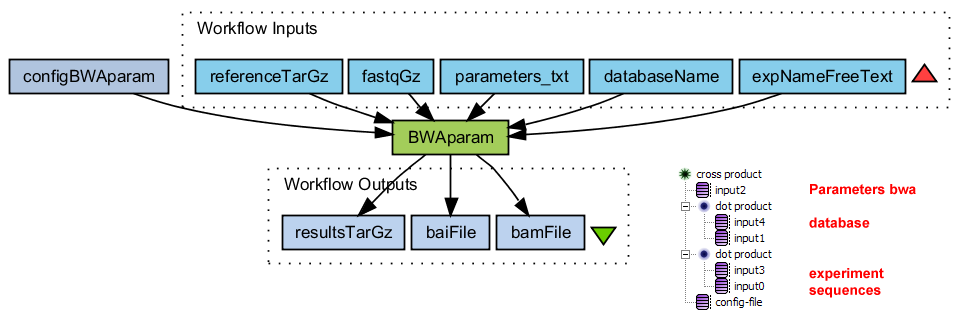
Alignment with BWA on each split file

Runs BWA with adjustable parameter settings.
- Matches sequence reads to a reference database
- Convert sai to sam
- Convert sam to bam
- Sort bam file
- Index sorted bam file
- Tar-gzip all results. Also the intermediate files
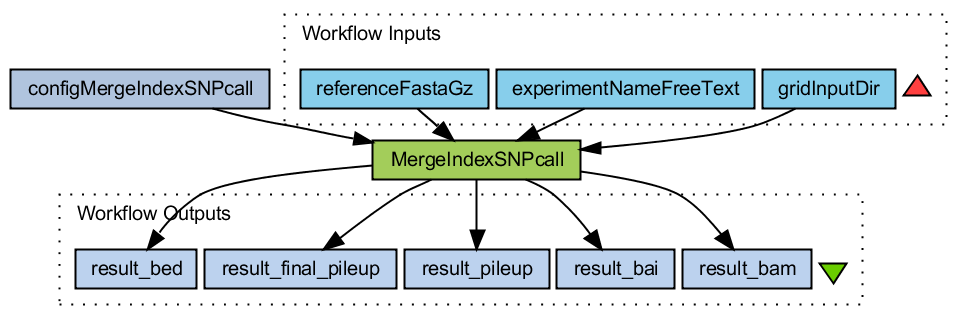
Merge bam files

- Downloads all bai, bam, sam and tar.gz files from the gridInputDirectory
- Gunzip tar the tar.gz files if they are present
- Gunzip the reference file (fasta format)
- Merge all _sorted.bam files
- Build index on this merged file
- Call SNPs and make selection. Output in pileup format.
- Convert pileup format to bed format
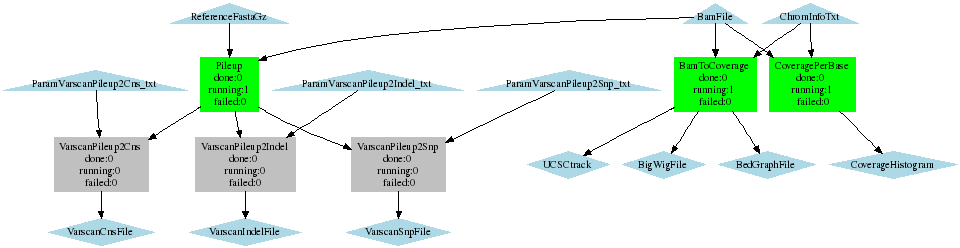
SNP calling with varscan, determine coverage

- Creates a pileup file (with samtools pileup -f) Sends the output to Varscan. Calls SNPs, indels and copy number variations.
- Calculates coverage per 50kbp
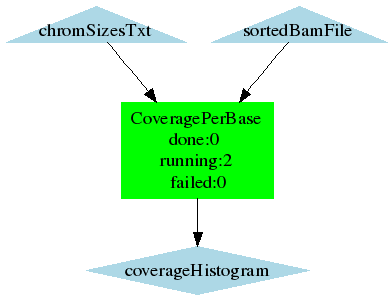
- Calculates coverage per base
Attachments (7)
-
CreateGridDirectory.png (2.9 KB) - added by 16 years ago.
WF CreateGridDirectory?
-
bwaIndexDatabase.png (44.7 KB) - added by 16 years ago.
WF Create BWA index on database
- splitFastq.png (31.0 KB) - added by 16 years ago.
-
BWAparam.png (39.1 KB) - added by 16 years ago.
WF Alignment with BWA
-
MergeIndexSNPcall.png (36.1 KB) - added by 16 years ago.
WF Merge bam files and call SNPs with samtools
-
Coverage_Varscan_BaseCoverage.png (8.8 KB) - added by 16 years ago.
WF call SNPs with varscan, calculate coverage per 50kb and per base
- CoveragePerBase.png (4.3 KB) - added by 16 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip