| Version 39 (modified by , 16 years ago) (diff) |
|---|
Table of Contents
SNP calling pipeline
Status: Alpha
Authors: Freerk van Dijk, Morris Swertz
Based on Broad GATK pipeline.
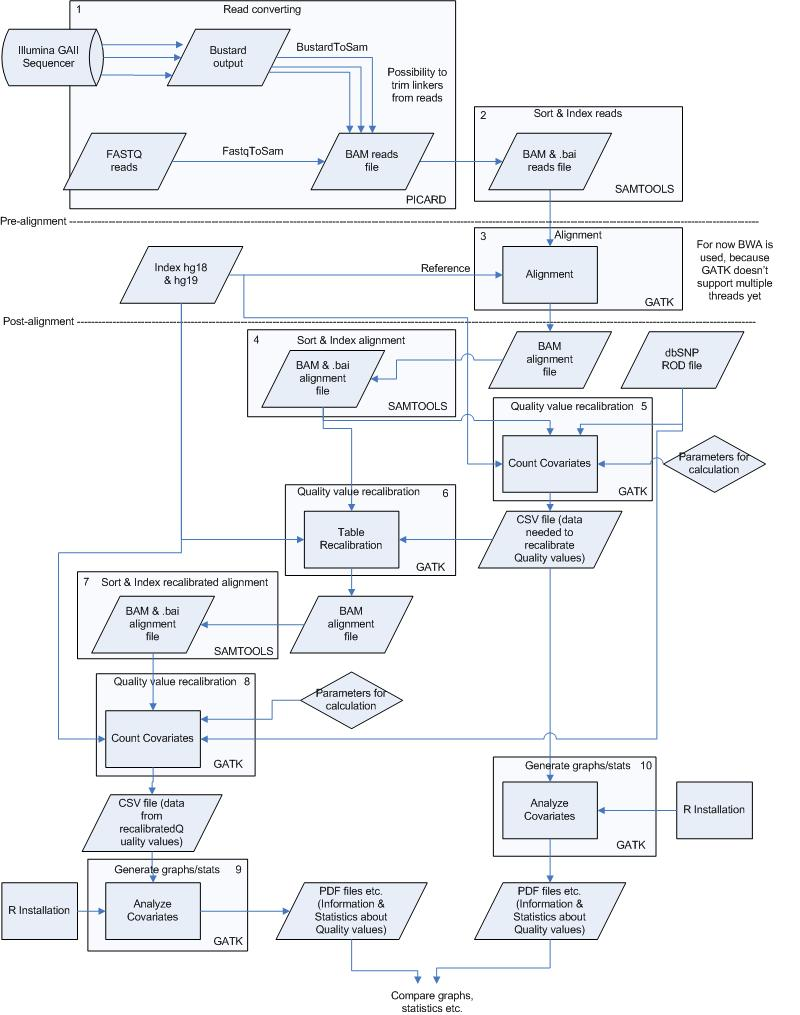
To perform the analysis as fast and good as possible the pipeline has been divided into several small processes. These processes are all numbered and can be found below, including commands, input and output files starting with pre-alignment and ending with variation calling & filtering.
Simplified Overview
This simplified overview this schema hides intermediate sort and indexing steps and only shows data inputs/outputs first time they occur.
Workflow 1: genome reference file creation
This workflow creates reference files per chromosome including:
- genome, dbsnp and indel vcfs per chromosome
- realign targets for faster realignment target creation
- index files for samtools and bwa
Workflow inputs:
- genome.chr.fa - downloaded from genome supplier (now hg19)
- dbsnpXYZ.rod - downloaded reference SNPs from dbsnp (now 129)
- indelsXYZ.vcf - downloaded reference indels from 1KG
Workflow outputs:
- genome.chr.fa - cleaned headers
- genome.chr.fa.fa - index for samtools
- genome.chr.fa.<format> - multilple index files for bwa
- dbsnpXYZ.chr.rod - split per chromosome
- indelsXYZ.chr.vcf - split per chromosome
- genome.chr.realign.intervals - targets for realignment
clean-fasta-headers
Clean headers to only have '1' instead of Chr1, etc
| tool: | |
| inputs: | genome.chr.fa |
| outputs: | genome.chr.fa |
| doc: | internally developed |
split-vcf-chr for dbsnp and indels
Split vcf per chromosome
| tool: | |
| inputs: | dbsnpXYZ.rod, indelsXYZ.vcf |
| outputs: | dbsnpXYz.chr.rod, indelsXYZ.vcf |
| doc: |
Discussion:
Can we use http://vcftools.sourceforge.net/options.html ?
vcftools --vcf indelsXYZ.vcf --chr <i> --recode --out indelsXYZ.chr
index-chromosomes
Index reference sequence for each chromosome in the FASTA format
| tool: | samtools faidx |
| input: | genome.chr.fa |
| output: | genome.chr.fa.fai |
| doc: | http://samtools.sourceforge.net/samtools.shtml#3 |
bwa-index-chromosomes
Index reference sequence for each chromosome for bwa alignment
| tool: | bwa index -a IS |
| input: | genome.chr.fa |
| output: | genome.chr.fa.xyz |
| doc: | http://bio-bwa.sourceforge.net/bwa.shtml#3 |
RealignerTargetCreator
Generate realignment targets for known sites for each chromosome
| tool: | GenomeAnalysisTK.jar -T RealignerTargetCreator? |
| input: | genome.chr.fa, dbsnpXYz.chr.rod, indelsXYZ.vcf |
| output: | genome.chr.realign.intervals |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels#Running_the_Indel_Realigner_only_at_known_sites |
Workflow 2: Alignment per Lane, per Chr
This workflow aligns reads per lane and chromosome, including:
- re-alignment to prevend false SNP calls caused by indels (using known indels)
- markduplicates to prevend false coverage caused by PCR errors (per library = lane)
- base quality recalibration to correct for false low scores caused by true variation
Workflow Inputs:
- lane.1.fq.gz - raw reads for lane, pair end 1
- lane.2.fq.gz - raw reads for lane, pair end 2
- genome.chr.fasta - reference genome split on chromosome
- genome.chr.realign.intervals - targets for realignment per chromosome
- genome.chr.dbsnpXYZ.rod - known snp variants, here from dpbsnp
- genome.chr.indelsXYZ.vcf - known indels from, here from 1KG
Workflow ouputs:
- lane.chr.1.sai - alignment index for first pair
- lane.chr.2.sai - alignment index for second pair
- lane.chr.sam - alignment map for
- lane.chr.bam - alignment map in binary format
- lane.chr.sorted.bam - sorted alignment map
- lane.chr.sorted.bai - sorted alignment index
- lane.chr.dedup.bam - marked duplicate PCR elements
- lane.chr.dedup.metrics - metrics describing deduplication
- lane.chr.realigned.bam - realigned based on known indels
- lane.chr.matefixed.bam - fixed the mate pair ends
- lane.chr.covariate_table.csv - table of countcovariates output for recalibration
- lane.chr.recal.bam - alignment map with recalibrated quality scores
align
Align each end of paired end.
| tool: | bwa-align |
| input: | chr.fasta, lane.1.fq.gz, lane.2.fq.gz |
| output: | lane.chr.1.sai, lane.chr.2.sai |
| docs: | http://bio-bwa.sourceforge.net/bwa.shtml |
align-pe
Align the pairs as one
| tool: | bwa sampe |
| inputs: | chr.fasta lane.1.fq.gz lane.2.fq.gz lane.chr.1.sai lane.chr.2.sai |
| outputs: | lane.chr.sam |
| docs: | http://bio-bwa.sourceforge.net/bwa.shtml |
sam-to-bam
Convert sam to bam
| tool: | samtools view |
| inputs: | lane.chr.sam |
| outputs: | lane.chr.bam |
| docs: | http://samtools.sourceforge.net/samtools.shtml |
(Question: can this not index and sort?)
sam-sort
Sort bam file on coordinate
| tool: | samtools sort |
| inputs: | lane.chr.bam |
| outputs: | lane.chr.sorted.bam |
| docs: | http://samtools.sourceforge.net/samtools.shtml |
sam-index
Index bam file for quicker access
| tool: | samtools index |
| inputs: | lane.chr.sorted.bam |
| outputs: | lane.chr.sorted.bai |
| docs: | http://samtools.sourceforge.net/samtools.shtml |
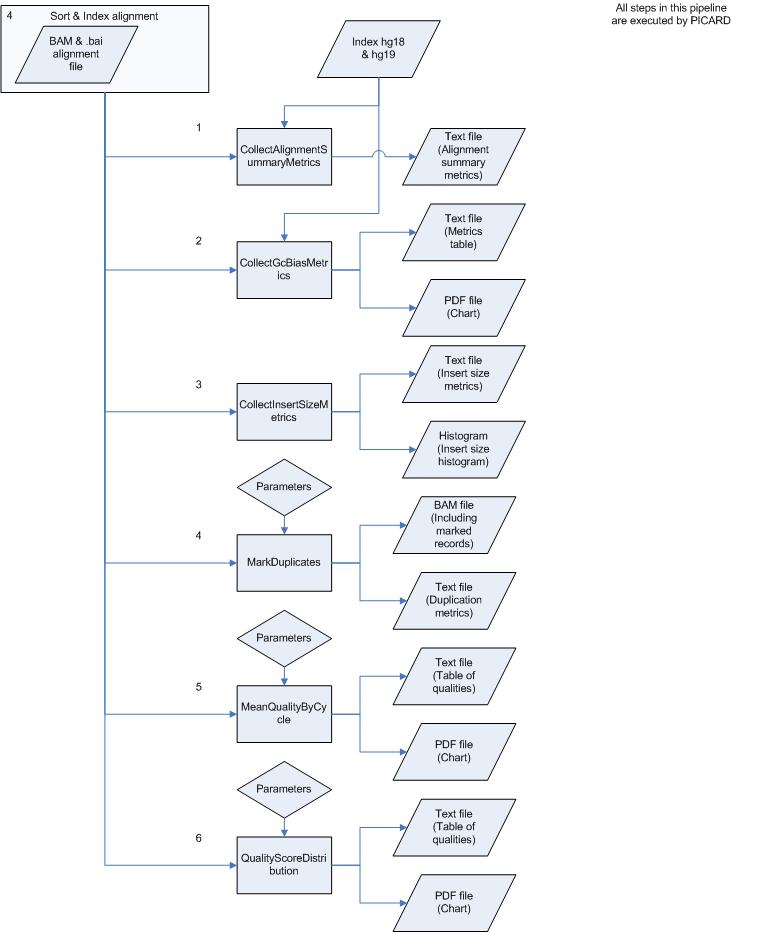
MarkDuplicates
Mark duplicate PCR fragments to be filtered in analysis
| tool: | MarkDuplicates?.jar |
| inputs: | lane.chr.sorted.bam |
| outputs: | lane.chr.dedup.bam lane.chr.dedup.metrics |
| docs: | http://picard.sourceforge.net/command-line-overview.shtml#MarkDuplicates |
IndelRealigner-KnownsOnly
Improve the alignment using known indel information (will reduce false SNP calls)
| tool: | GenomeAnalysisTK.jar -T IndelRealigner? |
| inputs: | lane.chr.dedup.bam genome.chr.realign.intervals genome.chr.dbsnpXYZ.rod genome.chr.indelsXYZ.vcf |
| outputs: | lane.chr.realigned.bam |
| docs | http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels#Running_the_Indel_Realigner_only_at_known_sites |
FixMateInformation
Fix the paired end information as consequence of the realignment.
| tool: | FixMateInformation?.jar |
| inputs: | lane.chr.realigned.bam |
| outputs: | lane.chr.matefixed.bam |
| docs: | http://picard.sourceforge.net/command-line-overview.shtml#FixMateInformation, |
CountCovariates
Count covariants, such as machine cycle and bp position, to be used as basis for quality recalibration. Optionally: plot the results to pdf using AnalyzeCovariates?
| tool: | GenomeAnalysisTK.jar -T CountCovariates?, AnalyzeCovariates?.jar |
| inputs: | lane.chr.matefixed.bam genome.chr.dbsnpXYZ.rod |
| outputs: | lane.chr.covariate_table.csv |
| docs: | http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration#CountCovariates |
TableRecalibration
Recalibrate quality scores based on the covariate table
| tool: | GenomeAnalysisTK.jar -T TableRecalibration? |
| inputs: | lane.chr.matefixed.bam lanec.chr.recal_table.csv chr.fasta |
| outputs: | lane.chr.recal.bam |
| docs: | http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration#TableRecalibration |
Repeat: sam-sort, sam-index, countcovariates
See steps above for commands and docs.
| inputs: | lane.chr.recal.bam |
| outputs: | lane.chr.recal.sorted.bam, lane.chr.recal.sorted.bam.bai, lane.chr.recal.covariate_table.csv |
Discussion:
wy do we need to sort and index after recalibration? does it mess up the order of things?
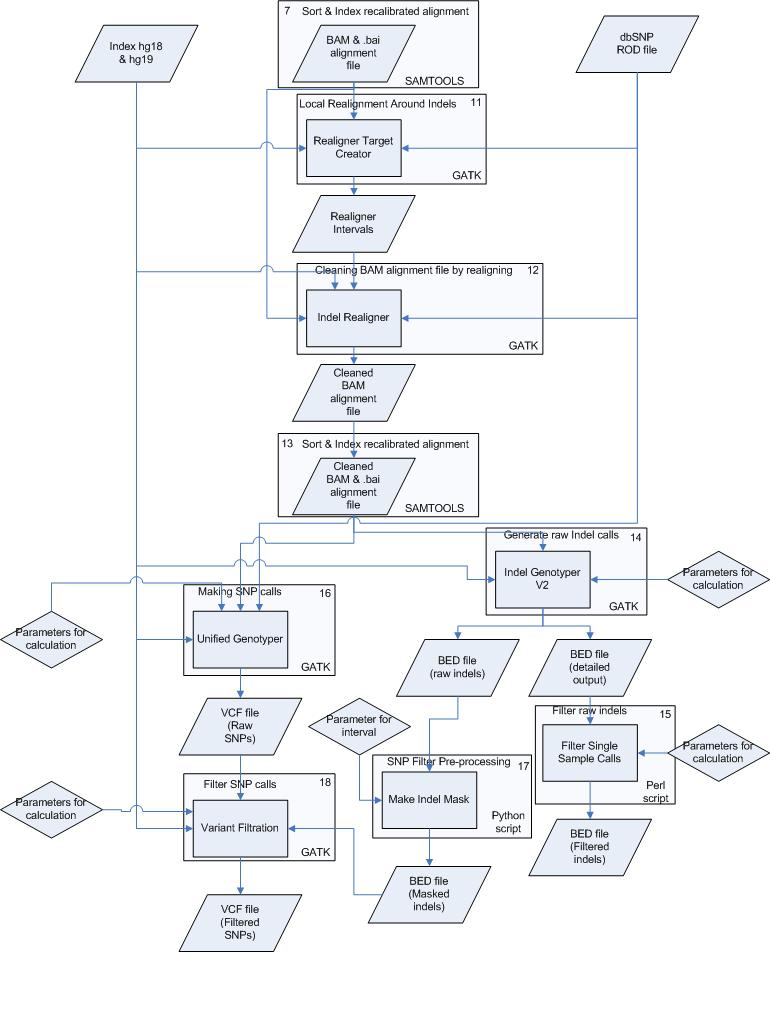
Workflow 3: sample level variant calling
This workflow will call variants for the samples including:
- sample level recalibration
- sample level realignment
N.B. no sample level MarkDuplicates? is needed as lanes == libraries.
Workflow inputs:
- lane.chr.recal.sorted.bam - for all sample lanes: dedupped, recalibrated, realigned, sorted and indexed bams (3)
- sample.chip.vcf - genotypes called from genotype chip
Reference:
- genome.chr.fasta - reference genome split on chromosome
- genome.chr.realign.intervals - targets for realignment per chromosome
- genome.chr.dbsnpXYZ.rod - known snp variants, here from dpbsnp
- genome.chr.indelsXYZ.vcf - known indels from, here from 1KG
Workflow outputs:
- sample.chr.bam - merged bam files per sample
- sample.chr.realign.interval - realignment target intervals
- sample.chr.realigned.bam - realigned
- sample.chr.matesfixed.bam - fixed pairs in realignment
- sample.chr.indels.vcf - raw indels called
- sample.chr.indels.bed - raw indels annotations
- sample.chr.indels.txt - output from the indel calling
- sample.chr.indels.filtered.bed - indels filtered
- sample.chr.snps.vcf - raw snps called
- sample.chr.snps.filtered.vcf - snps filtered
merge-lanes
Merge lanes into one sample bam
| tool: | sam merge |
| inputs: | lane.chr.recal.sorted.bam |
| outputs: | sample.chr.bam |
| docs: | http://samtools.sourceforge.net/samtools.shtml |
RealignerTargetCreator
Create realignment targets based on the data (so not only knowns)
| tool: | GenomeAnalysisTK.jar -T RealignerTargetCreator? |
| inputs: | sample.chr.bam genome.chr.fa dbsnpXYz.chr.rod indelsXYZ.vcf |
| outputs: | sample.chr.realign.intervals |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels#Creating_Intervals |
IndelRealigner
Realign based on realignment targets in previous step
| tool: | GenomeAnalysisTK.jar -T IndelRealigner? |
| inputs: | sample.chr.bam genome.chr.realign.intervals genome.chr.dbsnpXYZ.rod genome.chr.indelsXYZ.vcf |
| outputs: | sample.chr.realigned.bam |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels#Realigning |
FixMateInformation
See description in workflow2, now applied to sample
| inputs: | sample.chr.realigned.bam |
| ouputs: | sample.chr.matesfixed.bam |
IndelGenotyperV2
Call indels
| tool: | GenomeAnalysisTK.jar -T IndelGenotyperV2 |
| inputs: | sample.chr.matesfixed.bam genome.chr.fa |
| outputs: | sample.chr.indels.vcf sample.chr.indels.bed sample.chr.indels.txt |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Indel_Genotyper_V2.0 |
filterSingleSampleCalls
Filter indels
| tool: | filterSingleSampleCalls.pl |
| inputs: | sample.chr.indels.bed |
| outputs: | sample.chr.indels.filtered.bed |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Firehose_Parameters#SampleIndelGenotyper |
UnifiedGenotyper
Call SNPs
| tool: | GenomeAnalysisTK.jar -T UnifiedGenotyper? |
| inputs: | sample.chr.matesfixed genome.chr.fa dbsnpXYz.chr.rod |
| outputs: | sample.chr.snps.vcf |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Firehose_Parameters#SetUnifiedGenotypertoEval |
!makeIndelMask
Make indel mask
| tool: | makeIndelMask.py |
| inputs: | sample.chr.indels.bed |
| outputs: | sample.chr.indels.mask.bed |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Indel_Genotyper_V2.0#Creating_a_indel_mask_file |
VariantFiltration
Filter variants to get the best calls possible
| tool: | GenomeAnalysisTK.jar -T VariantFiltration? |
| inputs: | sample.chr.snps.vcf genome.chr.fa dbsnpXYz.chr.rod |
| outputs: | sample.chr.snps.filtered.vcf |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Best_Practice_Variant_Detection_with_the_GATK_v2#Integrating_analyses:_getting_the_best_call_set_possible |
MergeVcfs
ChipVcf
Produce vcf for the chips
VariantEval
Create summary information on the variations called for evaluation. Run per sample.snps.filtered.vcf against chip.
| tool: | GenomeAnalysisTK.jar -T VariantEval? |
| inputs: | sample.snps.vcf sample.chip.vcf genome.chr.fa dbsnpXYz.chr.rod |
| outputs: | sample.snps.eval |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/VariantEval |
Discussion:
Do we call SNPs based on the filtered indels or the raw indels? Should we realign AGAIN after merge of lanes? BAQ? MINDEL/PINDEL?
Attachments (3)
- Figure1.png (349.2 KB) - added by 16 years ago.
- Figure2.png (311.5 KB) - added by 16 years ago.
- Figure3.png (224.0 KB) - added by 16 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip