| Version 42 (modified by , 16 years ago) (diff) |
|---|
Table of Contents
SNP calling pipeline
Status: Alpha
Authors: Freerk van Dijk, Morris Swertz
Based on Broad GATK pipeline.
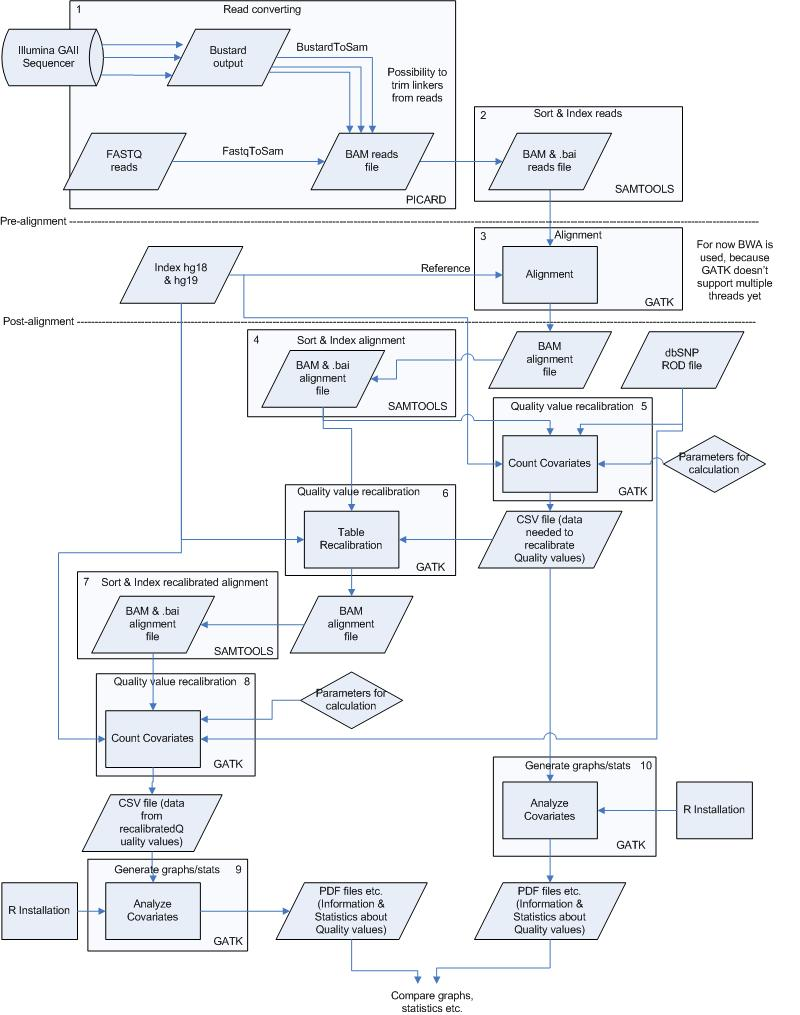
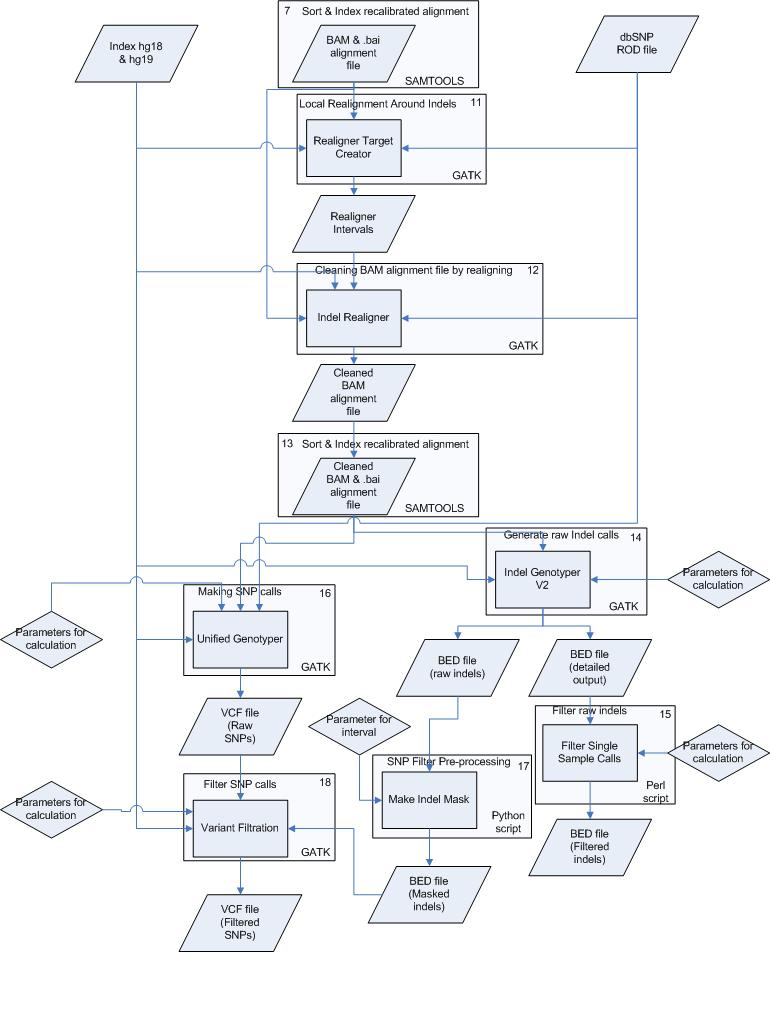
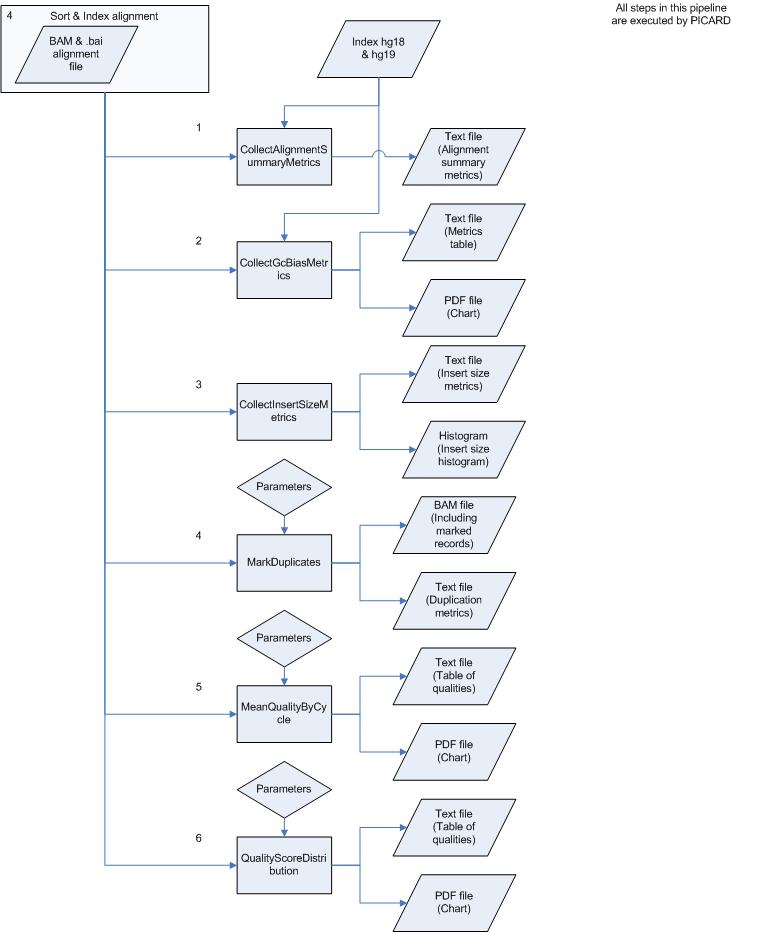
To perform the analysis as fast and good as possible the pipeline has been divided into several small processes. These processes are all numbered and can be found below, including commands, input and output files starting with pre-alignment and ending with variation calling & filtering.
- SnpCallingPipeline/ReferencePreparation
- SnpCallingPipeline/AlignmentAndCleaning
- SnpCallingPipeline/VariantCalling
Simplified Overview
This simplified overview this schema hides intermediate sort and indexing steps and only shows data inputs/outputs first time they occur.
Workflow 1: genome reference file creation
This workflow creates reference files per chromosome including:
- genome, dbsnp and indel vcfs per chromosome
- realign targets for faster realignment target creation
- index files for samtools and bwa
Workflow inputs:
- genome.chr.fa - downloaded from genome supplier (now hg19)
- dbsnpXYZ.rod - downloaded reference SNPs from dbsnp (now 129)
- indelsXYZ.vcf - downloaded reference indels from 1KG
Workflow outputs:
- genome.chr.fa - cleaned headers
- genome.chr.fa.fa - index for samtools
- genome.chr.fa.<format> - multilple index files for bwa
- dbsnpXYZ.chr.rod - split per chromosome
- indelsXYZ.chr.vcf - split per chromosome
- genome.chr.realign.intervals - targets for realignment
clean-fasta-headers
Clean headers to only have '1' instead of Chr1, etc
| tool: | |
| inputs: | genome.chr.fa |
| outputs: | genome.chr.fa |
| doc: | internally developed |
split-vcf-chr for dbsnp and indels
Split vcf per chromosome
| tool: | |
| inputs: | dbsnpXYZ.rod, indelsXYZ.vcf |
| outputs: | dbsnpXYz.chr.rod, indelsXYZ.vcf |
| doc: |
Discussion:
Can we use http://vcftools.sourceforge.net/options.html ?
vcftools --vcf indelsXYZ.vcf --chr <i> --recode --out indelsXYZ.chr
index-chromosomes
Index reference sequence for each chromosome in the FASTA format
| tool: | samtools faidx |
| input: | genome.chr.fa |
| output: | genome.chr.fa.fai |
| doc: | http://samtools.sourceforge.net/samtools.shtml#3 |
bwa-index-chromosomes
Index reference sequence for each chromosome for bwa alignment
| tool: | bwa index -a IS |
| input: | genome.chr.fa |
| output: | genome.chr.fa.xyz |
| doc: | http://bio-bwa.sourceforge.net/bwa.shtml#3 |
RealignerTargetCreator
Generate realignment targets for known sites for each chromosome
| tool: | GenomeAnalysisTK.jar -T RealignerTargetCreator? |
| input: | genome.chr.fa, dbsnpXYz.chr.rod, indelsXYZ.vcf |
| output: | genome.chr.realign.intervals |
| doc: | http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels#Running_the_Indel_Realigner_only_at_known_sites |
Attachments (3)
- Figure1.png (349.2 KB) - added by 16 years ago.
- Figure2.png (311.5 KB) - added by 16 years ago.
- Figure3.png (224.0 KB) - added by 16 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip